
(paper submitted to Bioinformatics)
The following project procedure is an example of how we used the WEKA machine learning toolkit, and one way you might want to use WetCat.
Objective: Create an interpretable classifier that can discriminate between HIV-1 sequences that use the CCR5 co-receptor exclusively and those that are capable of using the CXCR4 coreceptor. Essentially, given two sets of sequences from the same region of the HIV genome (V3), but with different (experimentally determined) class labels see if there is a consistent difference between them.
Basic Procedure:
Results: SVM classifies 91% of test data correctly. C4.5 classifies 89.5% of test data correctly and (with some attribute selection to limit the number of positions used) produces the following decision tree.
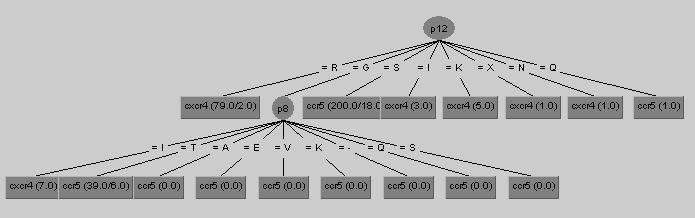
Which at the moment would look like this in the WetCat window:
J48 pruned tree ------------------ p12 = R: cxcr4 (61.0/2.0) p12 = G | p8 = I: cxcr4 (4.0) | p8 = T: ccr5 (26.0/6.0) | p8 = A: ccr5 (0.0) | p8 = E: ccr5 (0.0) | p8 = V: ccr5 (0.0) | p8 = K: ccr5 (0.0) | p8 = Q: ccr5 (0.0) | p8 = -: ccr5 (0.0) | p8 = S: ccr5 (0.0) p12 = S: ccr5 (145.0/17.0) p12 = I: cxcr4 (2.0) p12 = K: cxcr4 (3.0) p12 = X: cxcr4 (1.0) p12 = N: cxcr4 (1.0) p12 = Q: ccr5 (1.0) Number of Leaves : 16 Size of the tree : 18If translated into english, both of these representations would read "If there is an R at position 12, then the class for the sequence is CXCR4, otherwise if there is a G at position 12 and a I at position 8 then CXCR4, otherwise if there is a G at position 12 and a T at position 8 then CCR5 ..."